文 / 利刃君
微信ID / ziyuanliren666
全文共3928字,推荐阅读时间10分钟。
Autodock是一款开源的分子模拟软件,最主要应用于执行配体—蛋白分子对接。它由Scripps研究所的Olson实验室开发与维护,官方网址是http://autodock.scripps.edu/,目前最新的版本是AutoDock 4.2.6,包括AutoDock和AutoGrid两个模块,AutoDock软件包下载地址为http://autodock.scripps.edu/downloads/autodock-registration/autodock-4-2-download-page/,包括Linux、Mac OS和Windows版本以及源代码。
AutoDockTools是AutoDock对接的可视化程序,最新版本为MGLTools1.5.6,下载地址为http://mgltools.scripps.edu/downloads。
这里利刃君为大家带来Windows的软件安装程序,可由文末获取。其他版本大家可以复制上面的链接进行下载。
半柔性对接教程
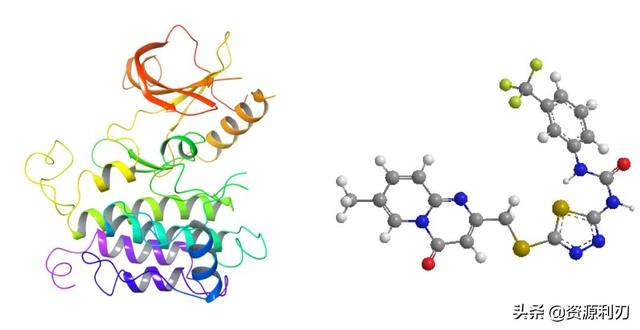
本次教程以蛋白6PL1的晶体结构作为分子对接的受体,晶体结构由蛋白数据库PDB网站下载,以其原配体OOJ为分子对接的配体,进行半柔性对接(对接受体蛋白设为刚性,对接配体小分子设为柔性)。
①可以使用pymol,VMD,Schrodinger,DS等软件,将蛋白晶体结构中的配体抽离出来,保存为Lig.mol2文件用于分子对接;
②可以使用pymol,VMD,Schrodinger,DS等软件将晶体结构6PL1中的水分子和原配体删除,保存为protein.pdb文件作为分子对接的受体。
下图中为处理好的蛋白晶体结构与配体结构。
-壹-
设置工作目录及工作环境
首先我们需要新建一个文件夹,设置该文件夹为我们的工作目录,此后所有分子对接产生的文件均默认保存在该文件夹中,方便我们进行查看。
注:该文件夹所有路径均要求为英文,不能出现中文和特殊字符。
利刃君这里在E盘新建文件夹“6PL1”,随后将下载好的“autogrid4,autodock4”程序以及“6pl1.pdb,Lig.mol2”两个文件拷贝到此文件夹下。
运行AutoDock Tool程序,选择File>Preferences>Set…>在Startup Directory下面的空里填入工作目录即刚才新建的文件夹路径“E:/6PL1”>Make Default
此后所有输入/输出文件的默认路径都是6PL1,即完成工作目录的设置。
-贰-
准备用于对接的受体
①导入蛋白:点击File>Read Molecule,选择6PL1文件夹中的“6pl1.pdb”,点击打开,导入蛋白文件。
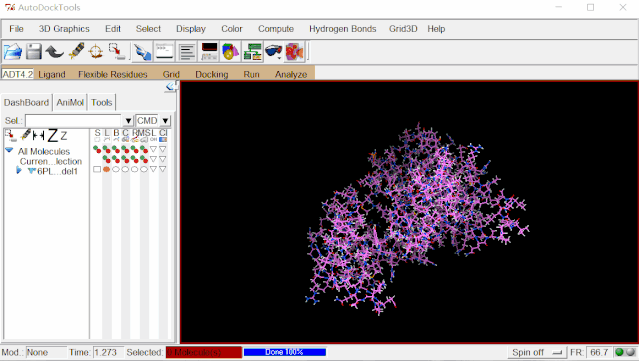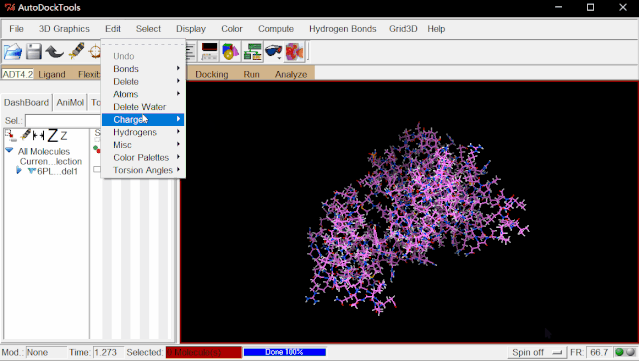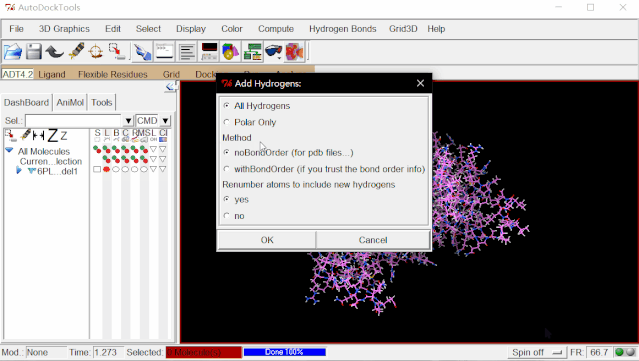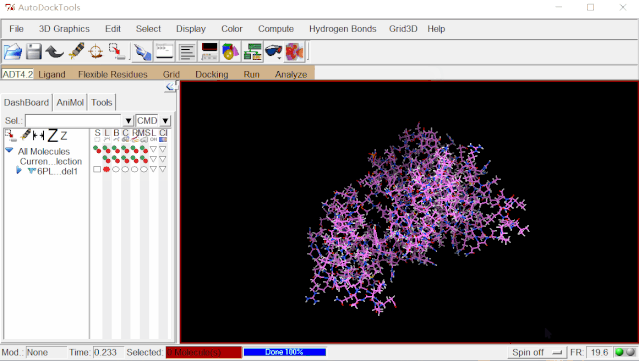
②加氢:点击Edit>Hydrogens>Add>点“OK”。(由于解析技术的原因,氨基酸的氢原子在晶体结构中是不存在的,因此需要手动加氢原子)
③加电荷:点击Edit>Charges>Compute Gasteiger>点“OK”。
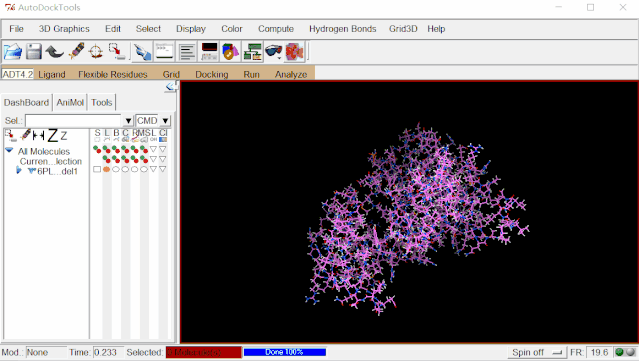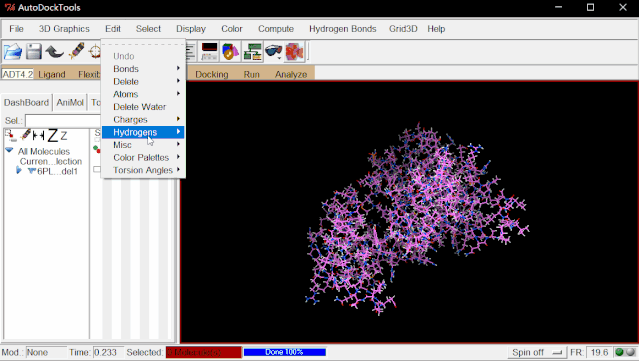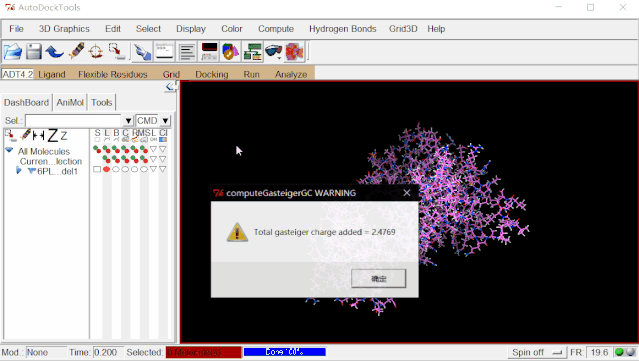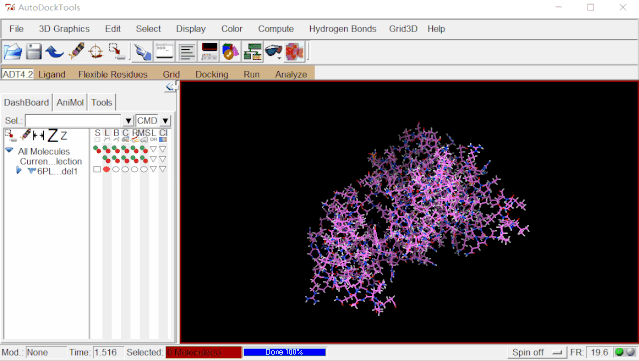
④保存处理好的蛋白文件:点击Grid>Macromolecule>Choose…>选择6pl1_model1后点“Select Molecule”>弹出的窗口点“确定”>弹出对话框点“保存”自动保存6pl1_model1.pdbqt。
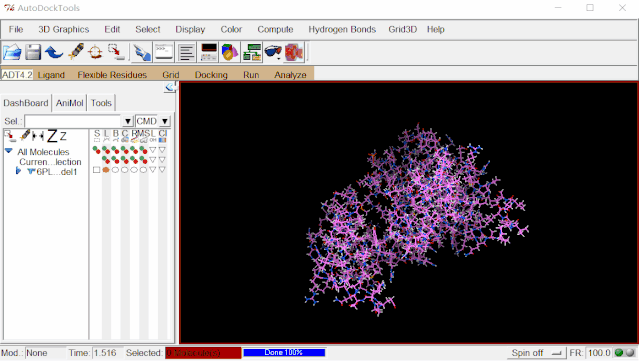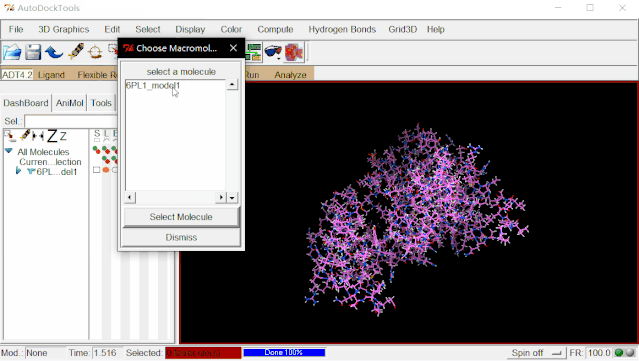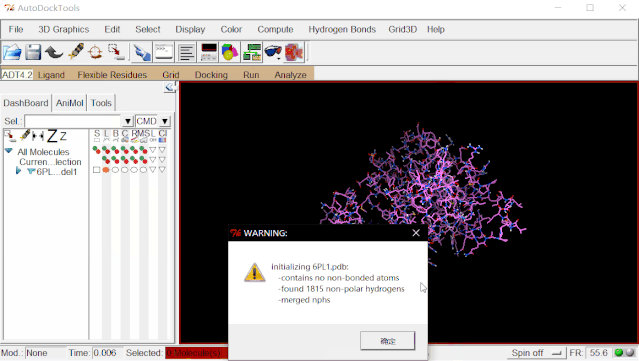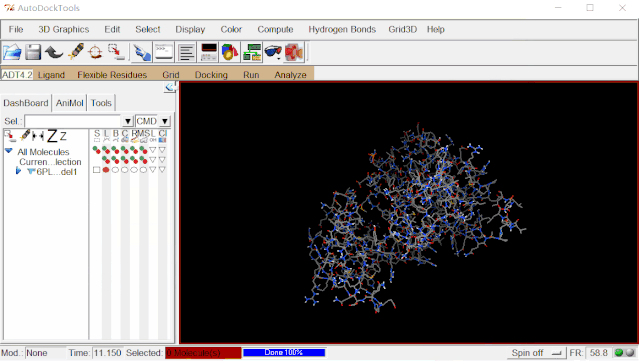
为避免打开多个分子导致有可能出现选择错误的问题,可点击Edit>Delete>Delete All Molecules>CONTINUE删除蛋白分子。
-叁-
准备用于对接的配体
对小分子配体进行加氢、加电荷(与蛋白的处理步骤相同)、加Root。
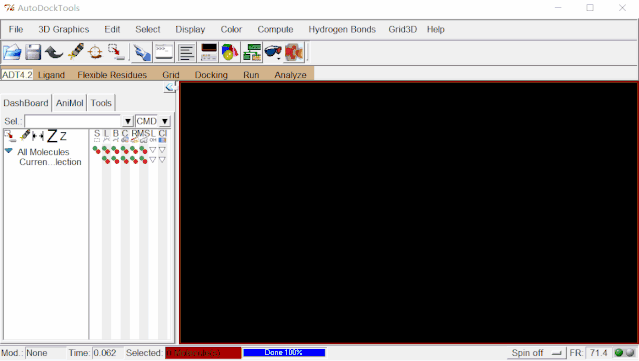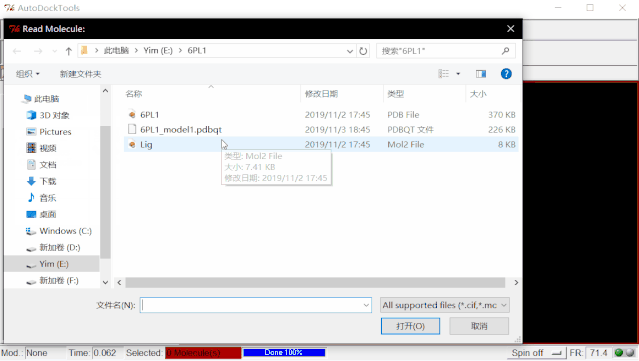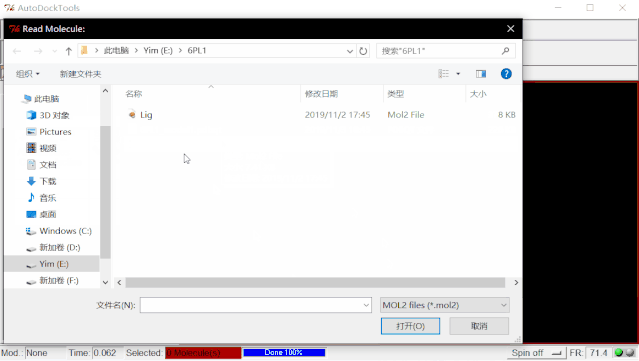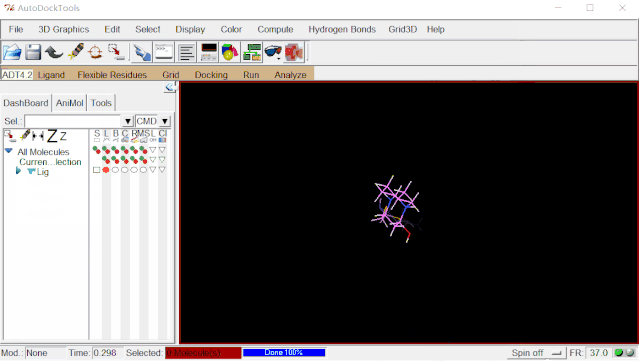
①点击File>Read Molecule,选择6PL1文件夹中的“Lig.mol2”,在打开的文件类型中选择mol2格式,点击打开,导入配体小分子。
②加氢:点击Edit>Hydrogens>Add>点“OK”。
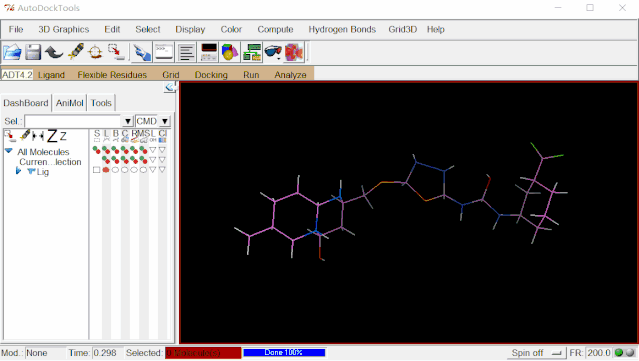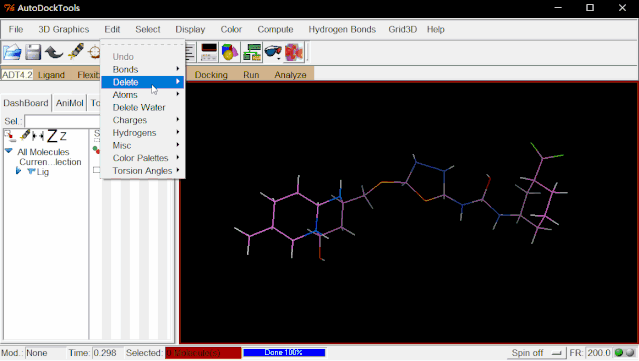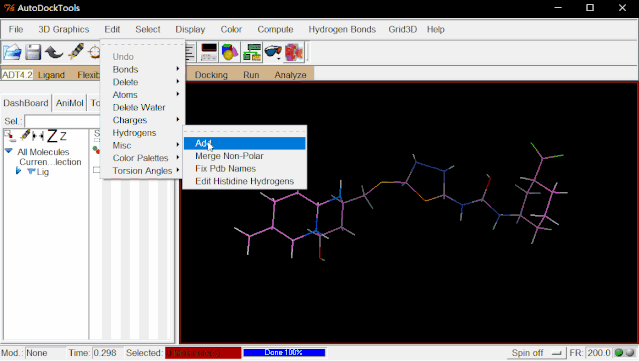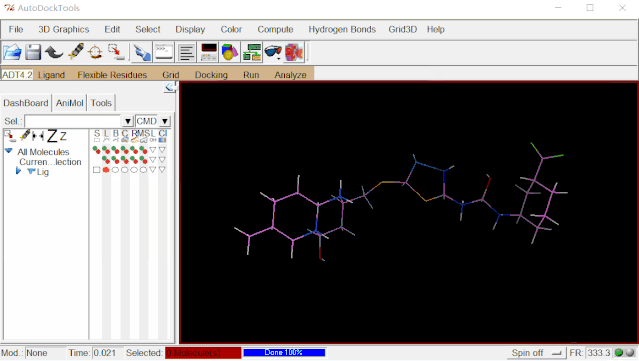
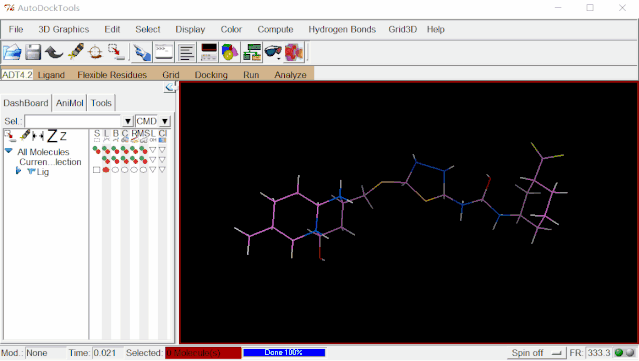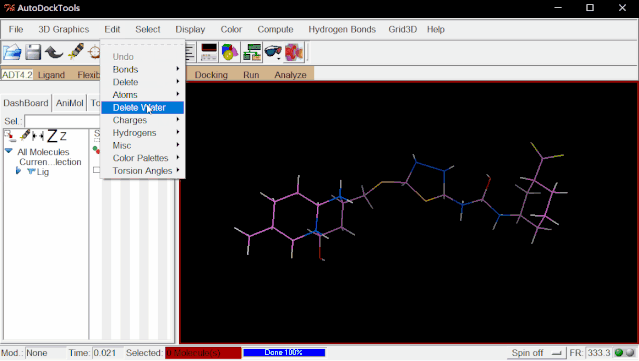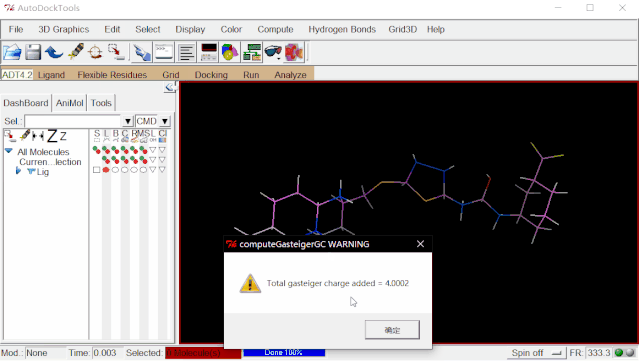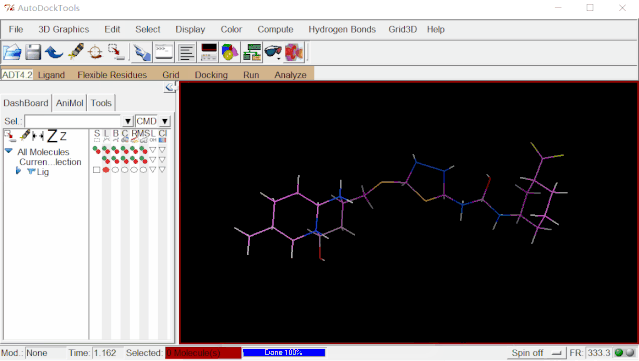
③加电荷:点击Edit>Charges>Compute Gasteiger>点“OK”。
④加Root:点击Ligand>Input>Choose…>选择Lig后点“Select Molecule for AutoDock 4”
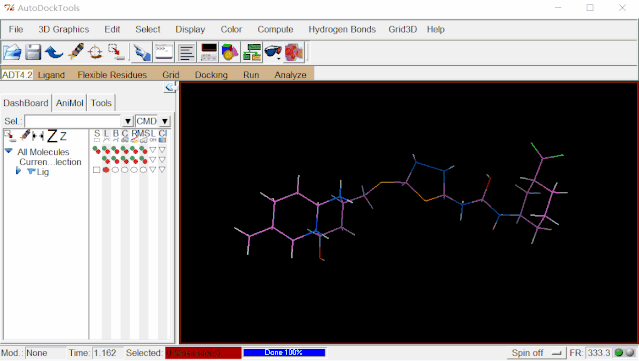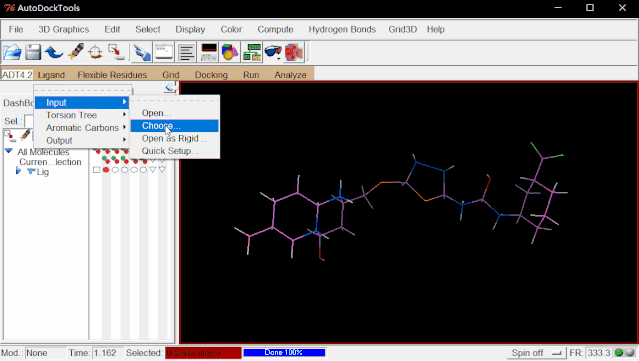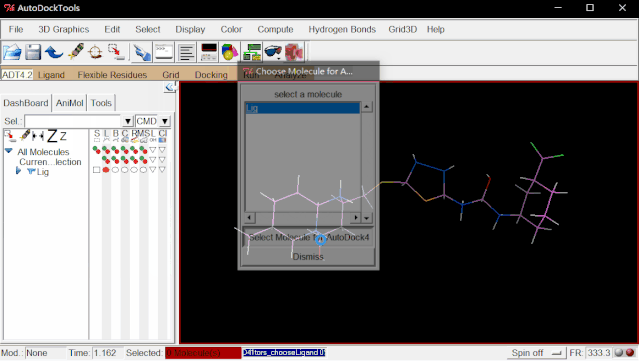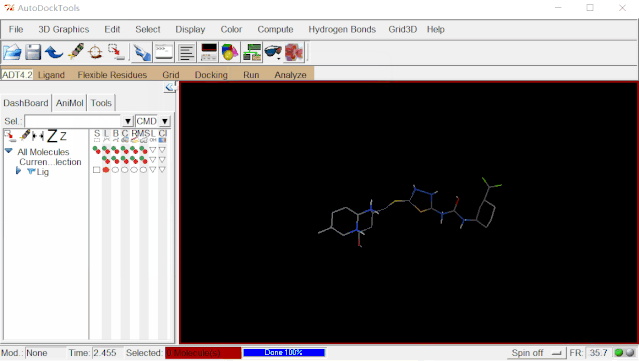
点击Ligand>Torsion Tree>Detect Root…(ADT自动判定Ligand的Root)
点击Ligand>Torsion Tree>Show Root Expansion(显示Root扩展信息)
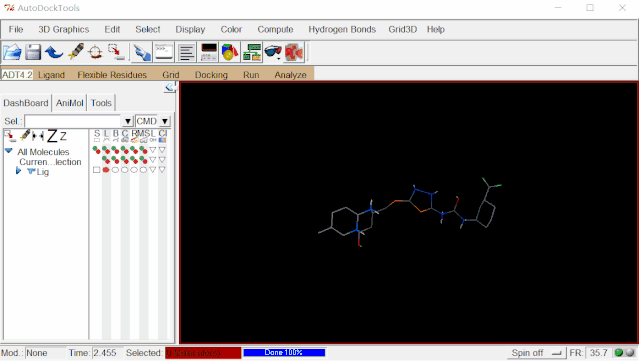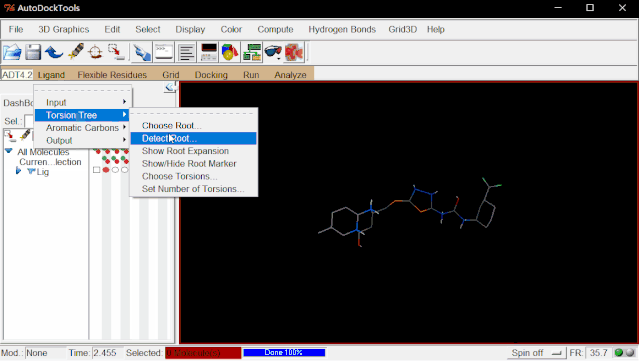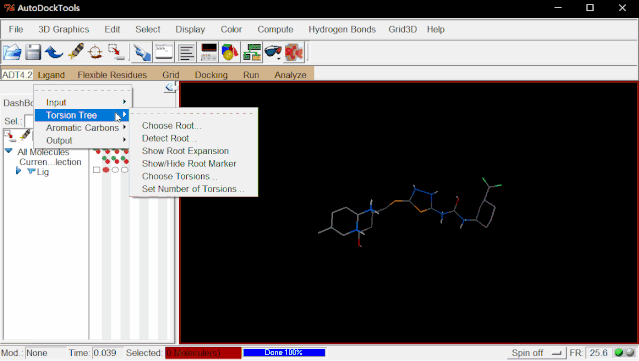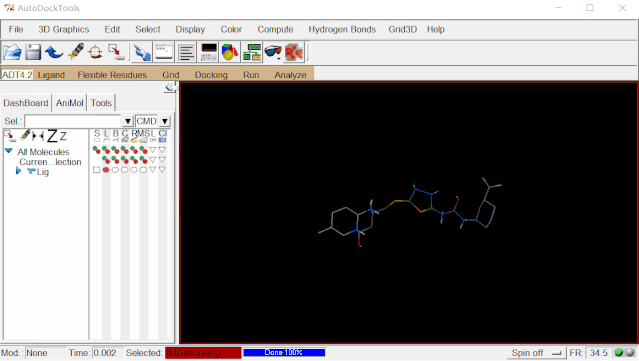
点击Ligand>Torsion Tree>Show/Hind Root Marker(显示/隐藏Root标记)
点击Ligand>Torsion Tree>Choose Torsions…>Done
点击Ligand>Output>Save as PDBQT…>保存
为避免打开多个分子导致有可能出现选择错误的问题,可点击Edit>Delete>Delete All Molecules>CONTINUE删除配体分子。
-肆-
进行Autogrid,生成受体网格
①导入受体:点击Grid>Macromolecule>Open…>6PL1_model.pdbqt>弹出的所有窗口点Yes/OK/确定。
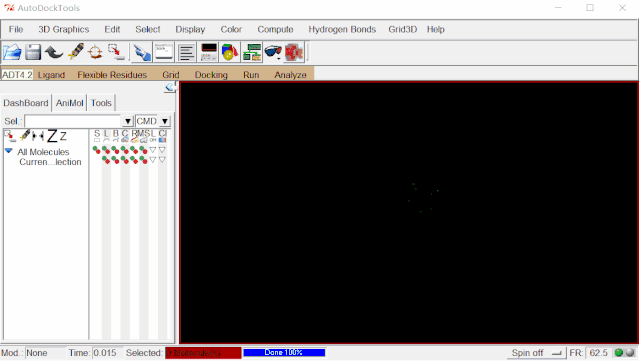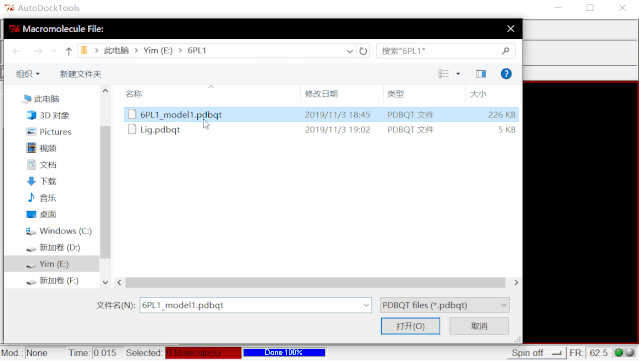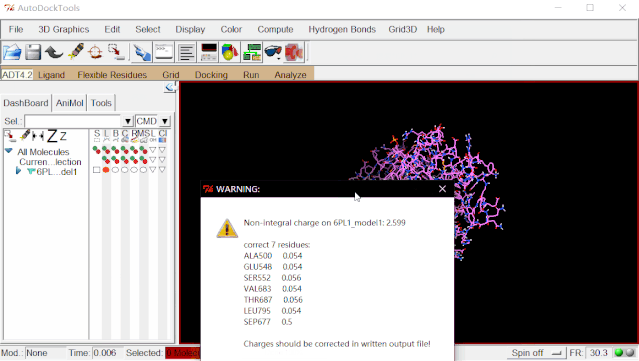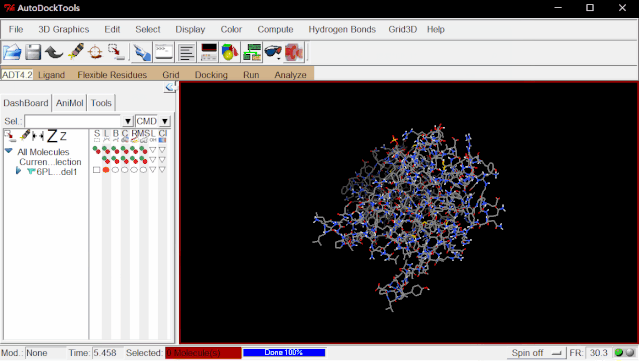
② 导入配体:点击Grid>Set Map Types>Open Ligand…>选择“Lig.pdbqt”>打开。
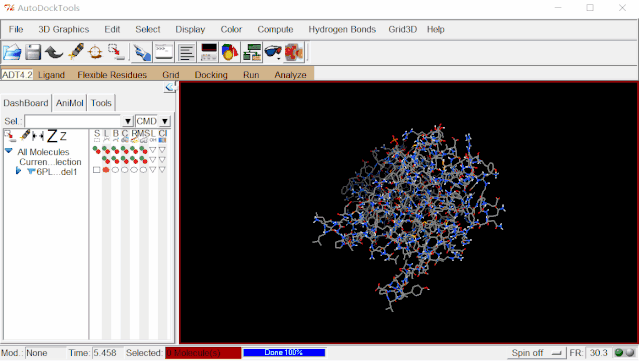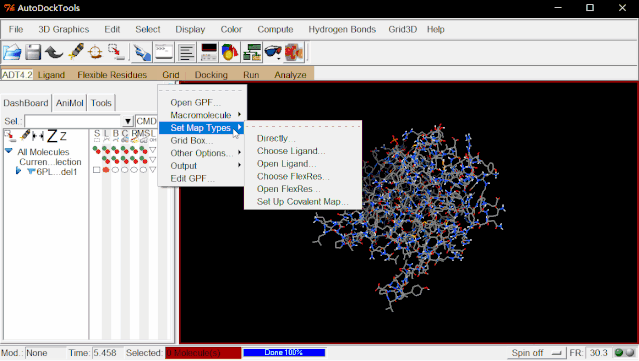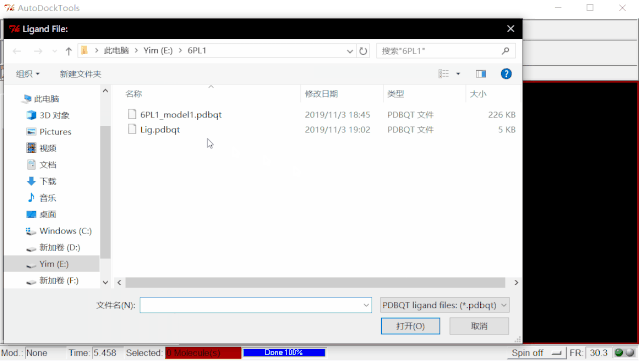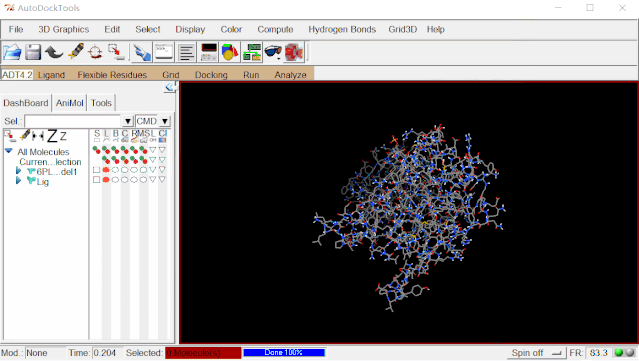
③生成受体网格:点击Grid>Grid Box设置对接的盒子大小,坐标,格点数,隔点距离,这一步需要自己根据不同的结构来进行具体确认(一般可查文献获取),对于有配体的蛋白我们往往通过配体扩张法来生成活性位点网格,最终目标是对接的盒子包含了配体可能结合的最大区域即可。
本文参数设置为x=40,y=40,z=74,Spacing=0.375,xyz分别表示在各方向上的格点的数量,Spacing表示每个格点的长度,盒子中心坐标为(-4.71,11.99,19.4),设置完成后可以在图形界面查看盒子的具体位置。
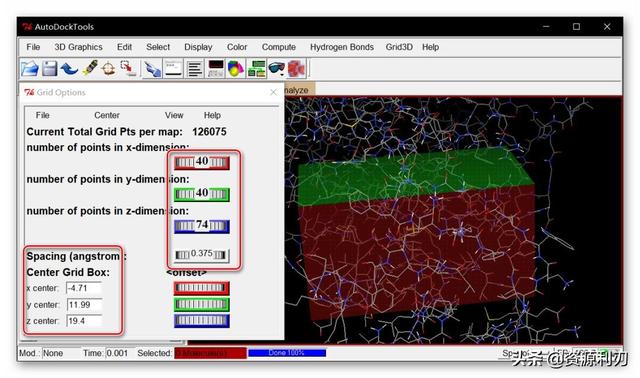
④保存网格文件:点击File>Close saving current保存盒子信息,选择Grid>Output>Save GPF,保存为protein_ligand.gpf文件(注意Windows下要手动添加文件名后缀)。
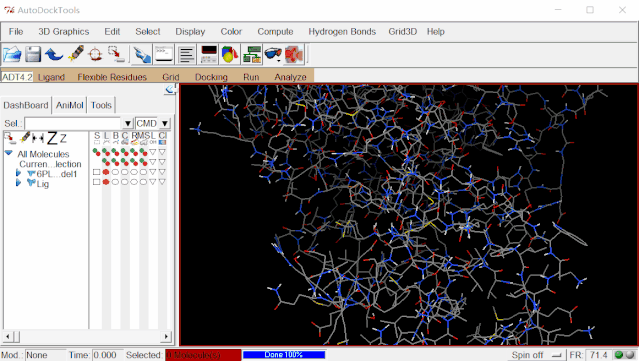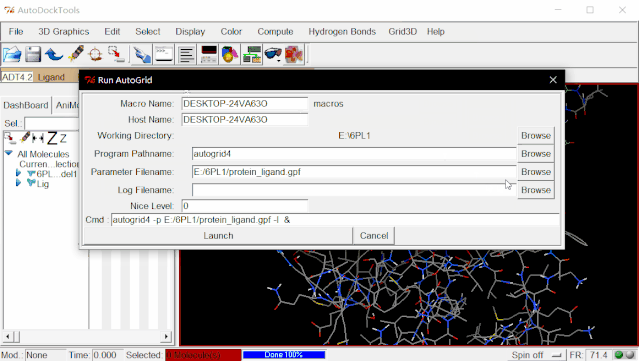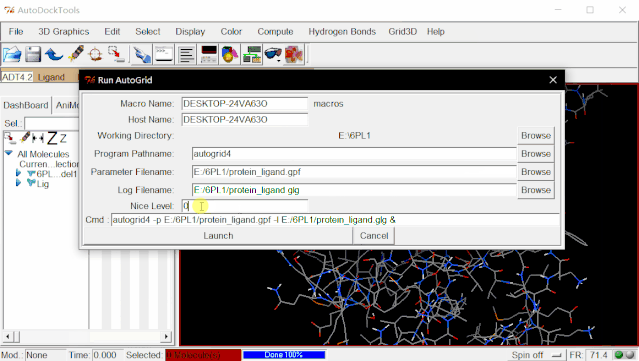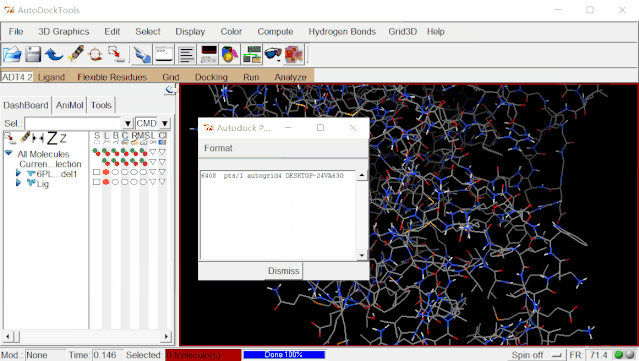
⑤进行AutoGrid:点击Run>Run AutoGrid,这里,单击Browse,在弹出来的页面里选择protein_ligand.gpf,点击打开,这样就自动生成了protein_ligand.glg文件。Nice Level设置为20(这里的值对对接结果没有什么影响),然后点击Launch运行。
AutoGrid4程序运行完毕后,除了生成一个protein_ligand.glg记录文件外,最主要的是生成一系列针对不同原子探针的范德华作用力、静电力以及去溶剂化作用力的Map文件,可以打开工作目录6PL1查看。
-伍-
进行分子对接
①设置对接受体:点击Docking>Macromolecule>Set Rigid Filename…>选择并打开6PL1_model.pdbqt,将受体蛋白质设置为刚性。

②设置对接配体:点击Docking>Ligand>Open…>打开Lig.pdbqt>Accept。
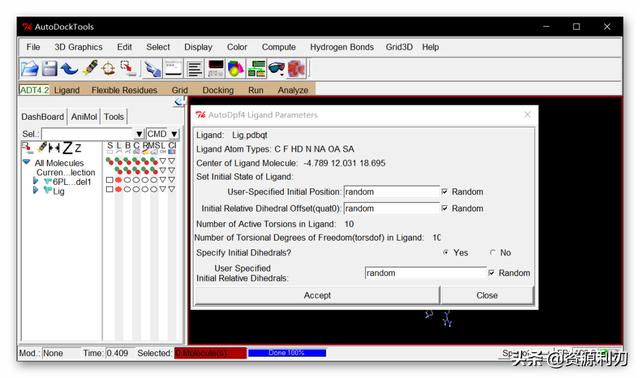
随后点击Docking>Ligand>Choose…>选择Lig,点“Select Ligand” 选择配体,设置初始位置等信息,点击Accept即可。
③设置对接信息:点击Docking>Search Parameters>Genetic Algorithm…>Accept
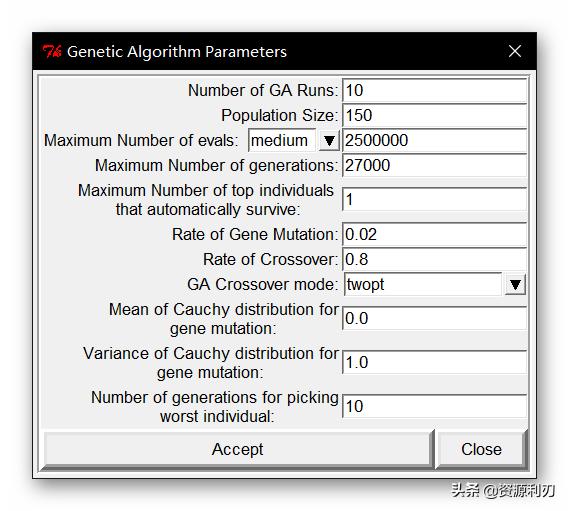
接着点击Docking>Docking Parameters…>Accept。
④保存文件:点击Docking>Output>Lamrckian GA 4.2,选择拉马克遗传算法作为对接算法,保存成为protein_ligand.dpf文件(Windows下手动加后缀),dpf文件中包含了分子对接的信息,默认对接的构象数为10个,可以用文本编辑器打开dpf文件,手动修改对接的构象数目(ga_run 10);

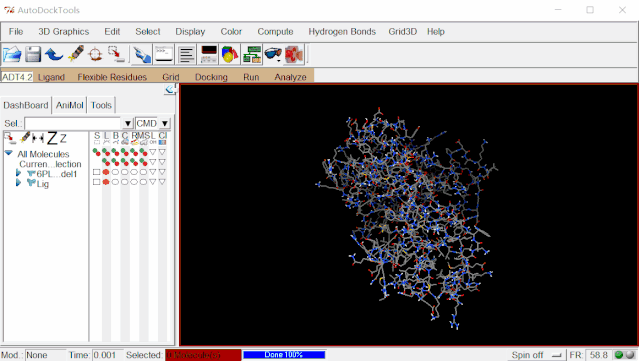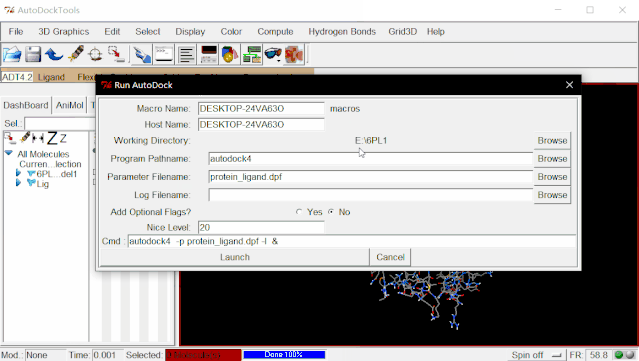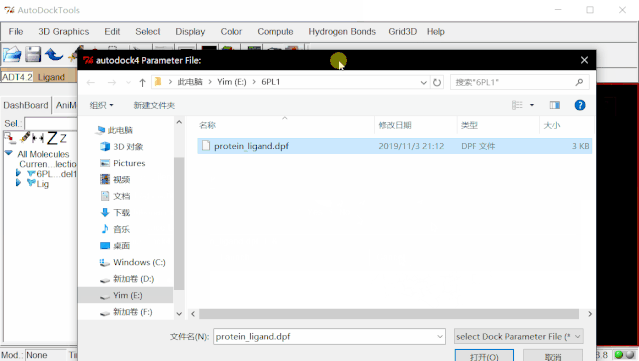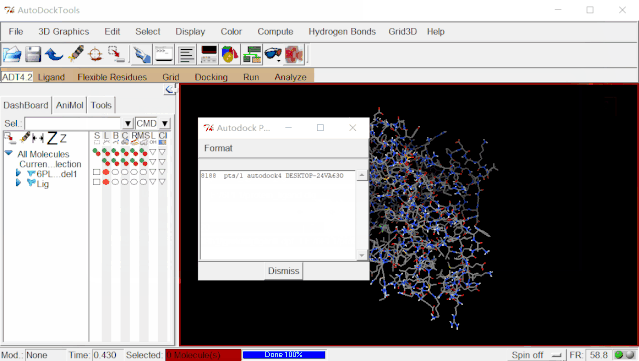
⑤进行分子对接:点击Run>Run AutoDock,在弹出的窗口中点击Browse,在弹出来的页面里再一次选择protein_ligand.dpf,点击打开,这样就自动生成了protein_ligand.dlg文件,Nice Level设置为20,然后点击Launch运行。
-陆-
查看对接结果
点击Edit>Delete>Delete All Molecules>CONTINUE删除配体分子。
点击Analyze>Dockings>Open…>选择并打开protein_ligand.dlg文件>出现的对话框都点Yes或确定。
之后点击Analyze>Conformations>Load…即可将对接结果及分子构象载入到图形窗口中,在弹出的protein_ligand Conformations Chooser对话框单击列表中对应的分子构象编号,上部的显示窗口即可显示该分子构象的诸如Binding Energy等分子对接信息。
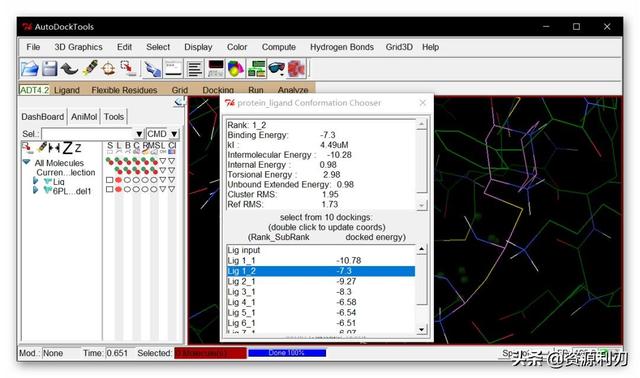
更多分析,比如聚类,构象叠合等大家可在analyze里找到。
资源获取方式:
1.点击头像,添加关注;
2.点击头像,私信关键词:036(注意关键词不要多字少字,否则后台无法识别)
就可获取Windows版本最新版AutoDock、AutoDock Vina、AutoDock Tool安装程序啦~