G:\r\tcga_example-master\scripts
library(survival)
library(survminer)## 批量生存分析 使用 coxph 回归方法
# http://www.sthda.com/english/wiki/cox-proportional-hazards-model
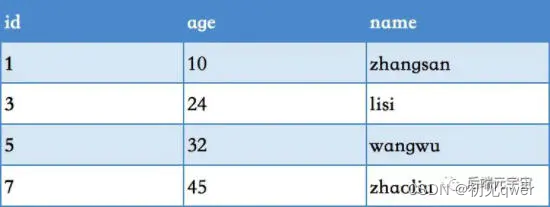
colnames(phe)
head(phe) #event中1 表示存活(终点事件)
#https://rpubs.com/kaz_yos/survival-auc
mySurv=with(phe,Surv(time, event)) # ## Create a survival vector
head(mySurv) #surv的结果只是一列一连串的生存时间汇总。所以一般会提倡用phe$mysurv这种方式把结果整合到原始数据框中便于之后的分析
#生存曲线与结果汇总
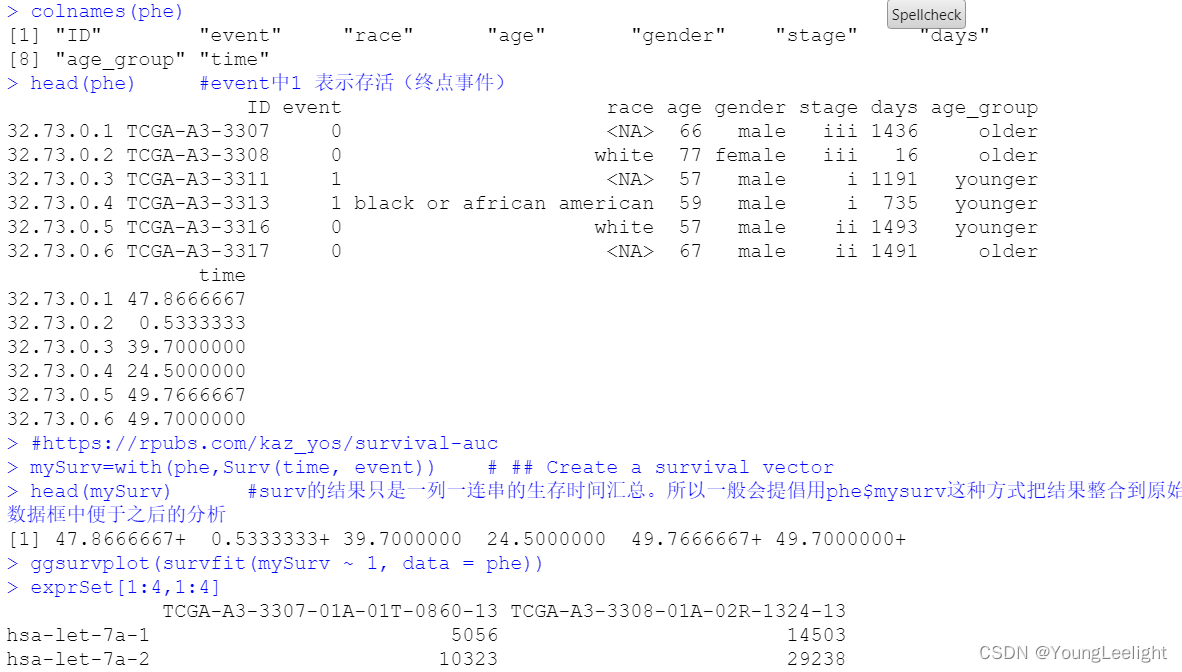
ggsurvplot(survfit(mySurv ~ 1, data = phe))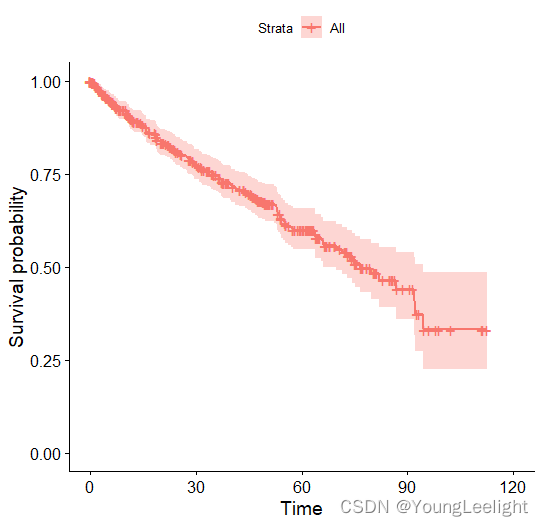
ggsurvplot(survfit(mySurv ~ age_group, data = phe), label.curves = list(method = "arrow", cex = 1.2))
library(survival)
data(pbc)
pbc <- within(pbc, {## transplant (1) and death (2) are considered events, and marked 1event <- as.numeric(status %in% c(1,2))## Create a survival vectorSurv <- Surv(time, event)
})
head(pbc)
ggsurvplot(survfit(Surv ~ sex, data = pbc), label.curves = list(method = "arrow", cex = 1.2))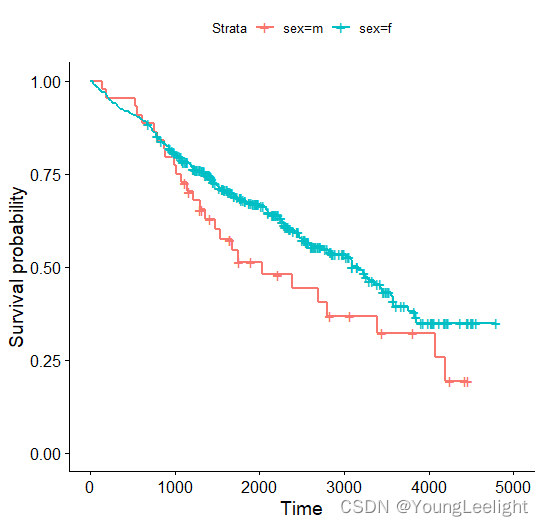
ggsurvplot(survfit(Surv ~ albumin >= median(albumin), data = pbc), label.curves = list(method = "arrow", cex = 1.2))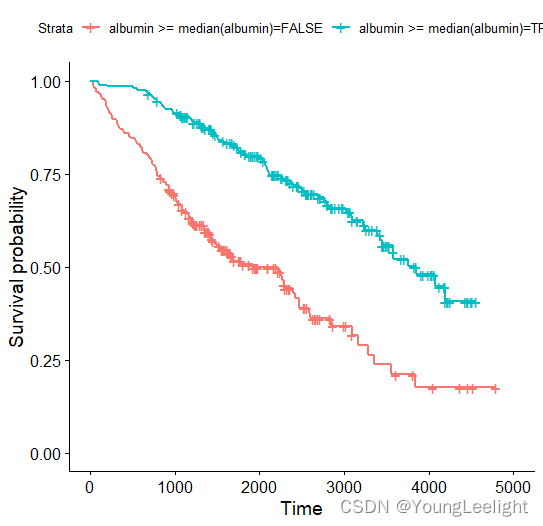
对五百多个miRNA基因进行批量运行cox,需要一点点时间。
如果是mRNA-seq的表达矩阵, 通常耗时更长。
注意,如果是某些基因表达量为恒定,比如在所有样本为0,这个代码会爆仓
需要去除这样的基因,没有分析的必要性。
exprSet[1:3,1:3]
#https://zhuanlan.zhihu.com/p/130316068
cox_results <-apply(exprSet , 1 , function(gene){
gene= exprSet[1,]
group=ifelse(gene>median(gene),‘high’,‘low’)
survival_dat <- data.frame(group=group,stage=phestage,age=phestage,age=phestage,age=pheage, #新建存活矩阵,试图阐明某个基因的高低表达对生存率的影响
gender=phe$gender,
stringsAsFactors = F)
m=coxph(mySurv ~ gender + age + stage+ group, data = survival_dat) #矫正其他因素
beta <- coef(m)
se <- sqrt(diag(vcov(m)))
HR <- exp(beta)
HRse <- HR * se
#summary(m)
tmp <- round(cbind(coef = beta, se = se, z = beta/se, p = 1 - pchisq((beta/se)^2, 1),
HR = HR, HRse = HRse,
HRz = (HR - 1) / HRse, HRp = 1 - pchisq(((HR - 1)/HRse)^2, 1),
HRCILL = exp(beta - qnorm(.975, 0, 1) * se),
HRCIUL = exp(beta + qnorm(.975, 0, 1) * se)), 3)
return(tmp[‘grouplow’,])
})
cox_results=t(cox_results)
head(cox_results)
table(cox_results[,4]<0.05)
cox_results[cox_results[,4]<0.05,] #coxph 回归方法得到p<0.05的所有miRNA, 接下来与logrank test 得到的结果对比一下
批量生存分析 使用 logrank test 方法
mySurv=with(phe,Surv(time, event))
log_rank_p <- apply(exprSet , 1 , function(gene){
gene=exprSet[1,]
phegroup=ifelse(gene>median(gene),′high′,′low′)data.survdiff=survdiff(mySurvgroup,data=phe)p.val=1−pchisq(data.survdiffgroup=ifelse(gene>median(gene),'high','low') data.survdiff=survdiff(mySurv~group,data=phe) p.val = 1 - pchisq(data.survdiffgroup=ifelse(gene>median(gene),′high′,′low′)data.survdiff=survdiff(mySurv group,data=phe)p.val=1−pchisq(data.survdiffchisq, length(data.survdiff$n) - 1)
return(p.val)
})
require(“VennDiagram”)
VENN.LIST=list(cox=rownames(cox_results[cox_results[,4]<0.05,]),#coxph 回归方法得到p<0.05的所有miRNA
log=names(log_rank_p[log_rank_p<0.05])) #logrank test 得到的p<0.05的所有miRNA
venn.plot <- venn.diagram(VENN.LIST , NULL,
fill=c(“darkmagenta”, “darkblue”),
alpha=c(0.5,0.5), cex = 2,
cat.fontface=4,
main=“overlap of coxph and log-rank test”)
可以看到两种生存分析挑出来的基因重合度尚可。
grid.draw(venn.plot)
save(log_rank_p,cox_results ,
file =
file.path(Rdata_dir,‘TCGA-KIRC-miRNA-survival_results.Rdata’)
)